我是一个生信菜鸟,在使用hisat2比对到基因组序列时出现了很多问题,下面简单说一下我踩过的坑。
1,个人觉得,除非要构建含有外显子和剪切位点的index,若只是比对到基因组上的话,可以直接从hisat2官网下载参考基因组的index,下载后会在一个文件夹中(人类的是hg19如果下载的hg19的,小鼠的是mm10),其中都是genome.123。。。.ht2文件
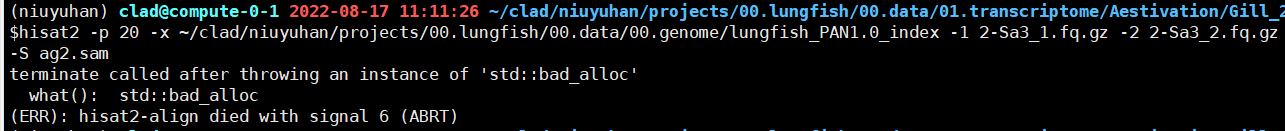
2,使用hisat2比对时,最好把index.x.ht2、.fq.gz文件、预存放.sam放到同一个文件夹下,不然可能会出错。。我也不知道为什么因为我很菜
3,使用hisat2之前一定要hisat2 -h看清其中的命令介绍,不然你会吃大亏! hisat2比对的一般参数如下
hisat2 –p 8 --dta –x hg19/genome -1 sample_1.fq.gz -2 sample_2.fq.gz –S sample.sam -p 8一般多少核去运行,这个看自己电脑的内存,我的16g选择八核稍微有点点卡,--dta是报告,-x是你的标识,hg19是你的index存放目录,而genome是你的index文件前缀!这时候插一句,如果你没有好好看清hisat2的要求,你可能就直接把hg19这个文件夹放到-x后面,或者将其中的genome.x.ht2利用正则表达式genome.*放到-x后面,再或者你可能利用cat > 整个成一个genome,这些都会导致hisat2不认识你的index文件,看清hisat2要求后,你就明白了,人家只是想要index的前缀,所以你只需把hg19/genome放到-x后面就可以!
后面的-1 -2的fastq文件一定要写对路径,或者就像我说的把他们放到同一个文件夹下,我喜欢用aligned命名文件夹
这样路径不会错,并且hisat2不会抽风的报错!
4,如果你的fastq文件很多,你可以在终端利用for...do...done写一个简单的循环for i in ’seq xx yy’ do hisat2 -p 8 - x hg19/genome(你的所有index文件前缀) -1 sample${i}1.fq.gz -2 sample${i}2.fq.gz -S sample${i}.sam
如果你想在vim中写一个sh的话,注意别忘了配置PATH
5,不会的一定要多查多问,不要闭门造車,不然很浪费时间!祝大家都成为生信高手!
最后附上一张运行图(我的Ubuntu就是那么可爱,还有小企鹅
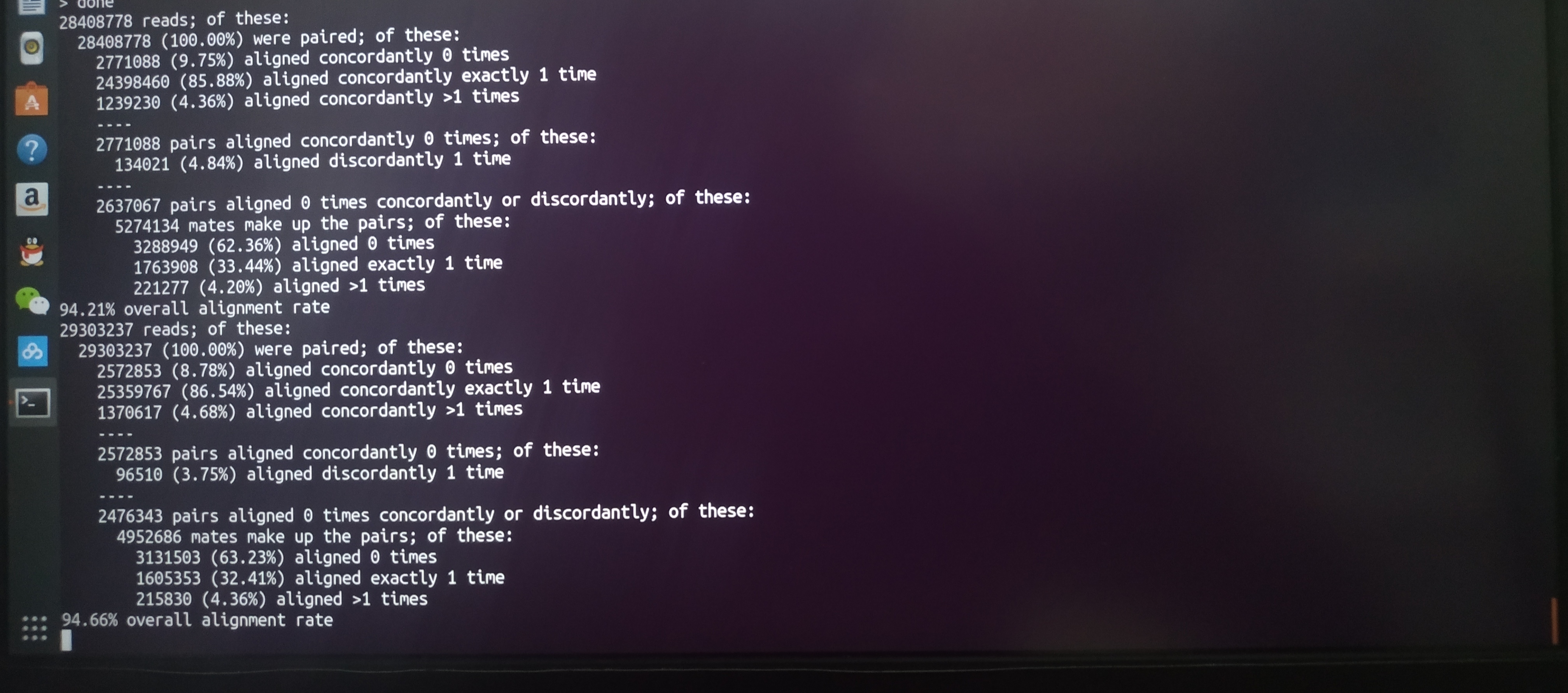
虽然这是centos的标签但是我还是想用哈哈)



 发帖
发帖 与我相关
与我相关 我的任务
我的任务
 分享
分享