社区
Power Linux
帖子详情
在Linux上没有办法下载hisat2,求各路大神帮忙看看
Shaquila_Chau
2021-01-15 11:12:40
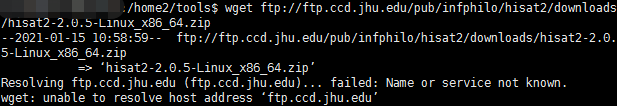
本人生物信息小白,对Linux环境还在摸索中,我在Linux服务器上下载不了hisat2,请问该如何解决?
我试过在网页上是可以打开网站的,但是在Linux上就说没有办法解释host address
求解答!谢谢!
...全文
3355
1
打赏
收藏
在Linux上没有办法下载hisat2,求各路大神帮忙看看
本人生物信息小白,对Linux环境还在摸索中,我在Linux服务器上下载不了hisat2,请问该如何解决? 我试过在网页上是可以打开网站的,但是在Linux上就说没有办法解释host address 求解答!谢谢!
复制链接
扫一扫
分享
转发到动态
举报
写回复
配置赞助广告
用AI写文章
1 条
回复
切换为时间正序
请发表友善的回复…
发表回复
打赏红包
h2plus0
2021-01-15
打赏
举报
回复
这个是 https://github.com/infphilo/hisat2 源码, 里面有Makefile, 一般输入 make 就可以编译了
hisat2
建立索引 序列比对rnaseq上游分析
linux
该博客聚焦RNA-Seq上游分析,推荐使用
HISAT2
进行序列比对。介绍了比对目的及不同工具适用场景,指出
HISAT2
在找新isoform等方面的优势。还说明了获取index的方法,以及将fastq比对得到sam文件,再用samtools转为bam文件、排序索引,对bam文件QC,最后用IGV可视化结果的流程。
【数据比对
hisat2
和samtools】零基础看懂运行代码、报错Q&A—结尾有彩蛋
本文详细介绍了如何在
Linux
环境下安装和使用
hisat2
进行RNAseq序列比对,以及如何利用samtools将比对结果转化为bam文件并进行排序。在遇到比对率低的问题时,作者提供了排查方法。此外,还讲解了如何通过IGV查看比对结果,以及使用featureCounts进行注释和计数,强调了基因组和注释文件来源的一致性。
Linux
下用
hisat2
手动去除RNA-seq中的rRNA:从NCBI
下载
到实战比对全流程
本文详细介绍在
Linux
环境下基于
HISAT2
手动去除RNA-seq数据中rRNA污染的完整生信流程:从NCBI
下载
RNA参考序列,筛选rRNA FASTA文件,构建
HISAT2
专用索引,执行比对并提取未比对读段(即非rRNA洁净数据),涵盖双/单端处理、跨物种适配及Shell脚本自动化。强调计算层面过滤原理、参数调优与质量评估,适用于提升下游定量准确性与分析效率。
老电脑也能跑生信:6G内存搞定RNA-seq全流程(附
Hisat2
+featureCounts实战代码)
本文面向低配置设备用户,详细阐述如何在仅6GB内存的老旧电脑上完成RNA-seq从SRA
下载
、质控、
HISAT2
比对到featureCounts定量的完整流程。重点涵盖
Linux
系统优化、swap配置、轻量软件选型、
HISAT2
内存精简参数(如--no-spliced-alignment)、featureCounts高效计数策略及OOM应急处理,并验证于小鼠转录组数据集,证明资源受限环境下仍可获得可靠表达矩阵。
HISAT2
高效比对实战指南:从安装到RNA-Seq数据分析(v2.2.1)
本文系统讲解
HISAT2
v2.2.1在RNA-Seq分析中的应用,涵盖
Linux
下三种安装方式(Conda/二进制/源码)、分层图FM索引构建(含剪接位点与SNP整合)、单双端数据比对命令及关键参数(--dta、--rna-strandness)、BAM直出优化技巧,以及比对摘要解读与常见问题排障。重点突出其高效性、剪接感知能力及生产环境最佳实践。
Power Linux
745
社区成员
900
社区内容
发帖
与我相关
我的任务
Power Linux
该论坛主要探讨Linux系统在IBM Power平台的安装、部署、应用开发等话题,并为网友们提供自由交流的平台。
复制链接
扫一扫
分享
社区描述
该论坛主要探讨Linux系统在IBM Power平台的安装、部署、应用开发等话题,并为网友们提供自由交流的平台。
社区管理员
加入社区
获取链接或二维码
近7日
近30日
至今
加载中
查看更多榜单
社区公告
暂无公告
试试用AI创作助手写篇文章吧
+ 用AI写文章



 发帖
发帖 与我相关
与我相关 我的任务
我的任务
 分享
分享

